The SIRIUS command-line tool can be called via the “binary/startscript” by simply running the command in the commandline:
sirius --help
TIP: We recommend using the --help option to get an overview of the available commands and options.
This ensures that the command descriptions are accurate and match your specific version of SIRIUS.
Introduction
The SIRIUS command-line program is a versatile toolbox designed for metabolite identification, offering a variety of tools (subcommands) that can be concatenated as toolchains to perform multiple analysis steps in a single run. The subcommands are categorized into different types:
- CONFIGURATION TOOL: The
configtool can be executed before any toolchain or standalone tool to configure all settings available in SIRIUS from the command line. - STANDALONE TOOLS: These tools operate independently and cannot be concatenated with other subtools. They are typically used for data management tasks, such as modifying project-spaces or exporting MGF files.
- PREPROCESSING TOOLS: Tools that prepare input data to be compatible
with SIRIUS. For example,
lcms-alignis used for feature detection and alignment. - COMPOUND TOOLS: These tools analyze each compound (instance) in
the dataset individually and can be concatenated with other tools. Examples are molecular formula annotation (
formulas), structure database search (structures) or compound class prediction (classes). - DATASET TOOLS: These tools analyze all compounds (instances) in
the dataset simultaneously and can be concatenated with other tools. For example, dataset-wide molecular formula annotation with
zodiac.
Each subtool can be called with the --help option to view the documentation
on available options and potential follow-up commands in a
toolchain. For example, to get help for the formulas tool, use:
sirius formulas --help
Basic workflow principles
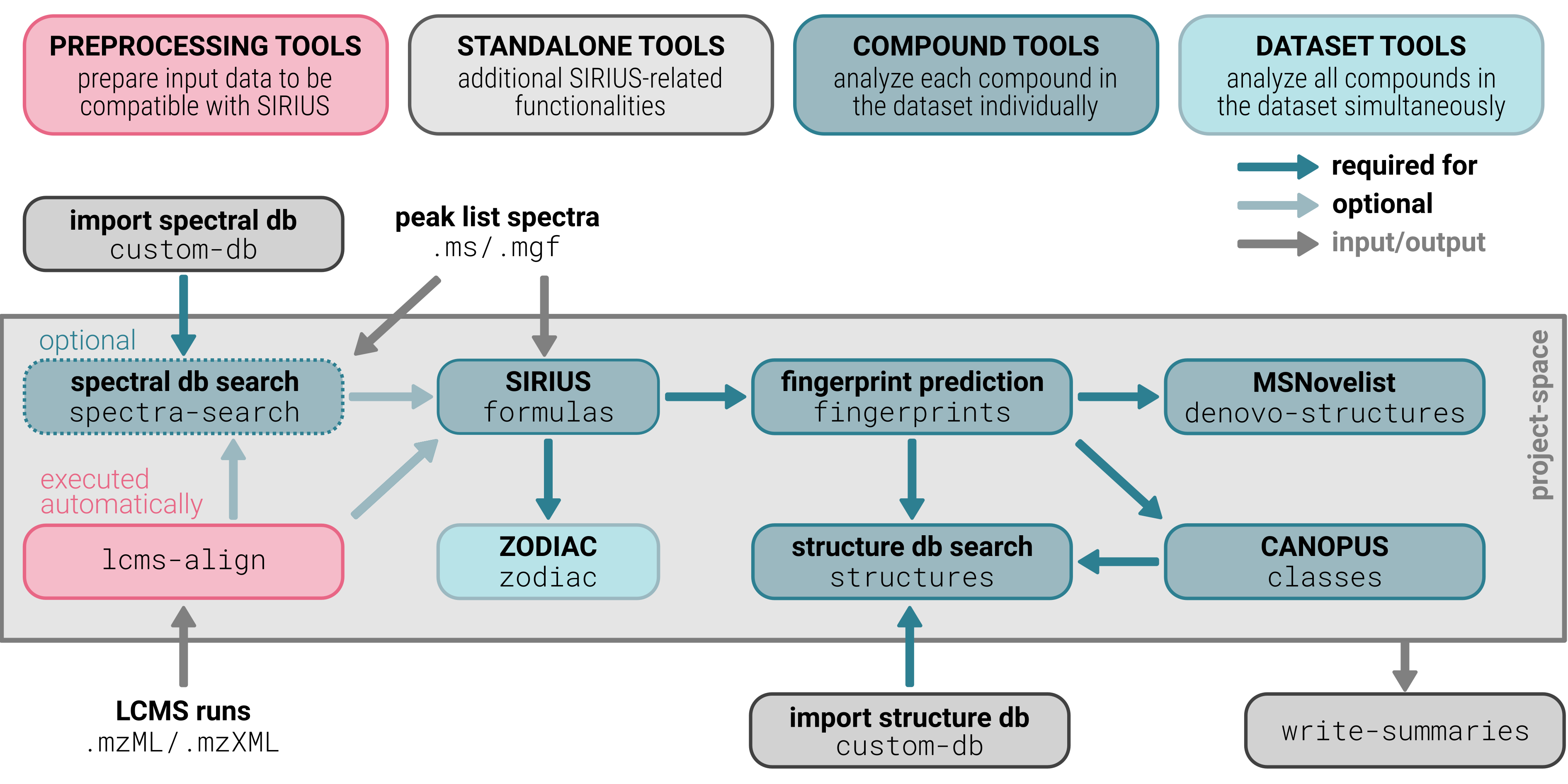
The SIRIUS CLI toolbox functions as a basic workflow engine (to generate toolchains), adhering to the following principles:
- Only the subtools explicitly specified in the command will be executed.
- Once the first subtool has been executed, a project-space is created. Subsequent commands will be executed on this project-space.
- If a mandatory input from a previous step (subtool) is missing, the computation for the current compound will be skipped.
- By default, the toolbox does not override existing results. Compounds for which results are already available will be skipped.
- If the
--recomputeoption is specified, existing results will be replaced with new ones for all subtools that are specified in the command. - When results for one subtool are recomputed (
--recompute), the results of downstream subtools that depend on recomputed results will be deleted, to ensure that all results remain consistent with the newly computed data. - Results from all subtools are stored in the project-space which can be visualized, modified, or further analyzed in the SIRIUS GUI.
Input, Project-Spaces, and Output
There are three different cases for importing spectra (we highly recommend using option 2 or 3):
- Importing spectra from generic text or CSV files on per compound level
- Importing multiple compounds from
.msor.mgffiles - Importing full LCMS runs from
.mzMLor.mzXMLfiles
The import is not a subtool and hence requires at least one of the subtools to be executed on the imported data. For case 3, lcms-align must be executed as first subtool. For cases 1 and 2, either formulas or spectra-search must be executed as first subtool. Once the first subtool has been executed, a project-space (--project <projectspace>) is created. Subsequent commands will be executed on this project-space. All results are saved in the project-space. From the project-space you can write summary files (output) into a custom location.
Example:
sirius --input <inputFile> --project <projectspace> formulas
sirius --project <projectspace> fingerprints
sirius --project <projectspace> write-summaries --output <location>
This can also be executed in a single command:
sirius --input <inputFile> --project <projectspace> formulas fingerprints write-summaries --output <location>
Importing spectra from generic text or CSV files on per compound level
sirius -1 <ms1File> -2 <ms2Files (comma separated)> -z <parentmass> --adduct <adduct> --project <projectspace> formula
ms1File is the MS1 spectrum containing the isotope pattern, and MS/ms2Files are the MS/MS fragmentation spectra. You can provide multiple MS/MS files (comma separated) if you have several measurements of the same compound with different collision energies; SIRIUS will merge these spectra into a single spectrum. Adduct nomenclature is described here. If you omit the --adduct option, [M+?]+ is used as default.
The command also works for MGF files, where you can omit the -z option for specifying the parent mass, if it is already given in the file.
Either formulas or spectra-search must be executed as first subtool.
Importing multiple compounds from .ms or .mgf files
We recommend using input files in .ms or .mgf format,
which contain all spectra for a compound as well as metainformation, such as parent mass, ionization and MS level. SIRIUS will extract the meta information from
the files. They can also contain multiple compounds per file.
sirius --input <inputFile> --project <projectspace> formulas
If you specify a directory instead of a single file, SIRIUS searches the directory for supported files, allowing for batch processing of multiple compounds.
Either formulas or spectra-search must be executed as first subtool.
Importing full LCMS runs from .mzML or .mzXML files
See lcms-align.
Filtering
You can filter features by precursor mass range, retention time range, data quality or the presence of MS/MS spectra. The filter does not apply for import but only to the subsequently applied tools.
sirius --input <inputFile> --project <projectspace> --<filteroption> formulas
Filter options are:
--mzmin=<mzmin>and--mzmax=<mzmax>for precursor mass range--rtmin=<rtmin>and--rtmax=<rtmax>for retention time range--hasmsmsto use only features with MS/MS data available--quality=<quality>[,<quality>...]to filter by data quality (LOWEST,BAD,DECENT,GOOD,NOT_APPLICABLE)
PREPROCESSING TOOLS
lcms-align: Feature detection and feature alignment (Preprocessing)
The lcms-align tool enables the import of multiple .mzML/.mzXML files into SIRIUS. It performs feature detection and alignment based on MS/MS spectra, creating a SIRIUS project-space, which can then be used for subsequent analysis steps. lcms-align is automatically executed when running the formulas tool on .mzML/.mzXML files:
sirius --input <mzml(s)> --project <projectspace> formulas
If you want to perform feature detection without alignment, you need to use the lcms-align tool with the --no-align option:
sirius --input <mzml(s)> --project <projectspace> lcms-align --no-align formulas
COMPOUND TOOLS
spectra-search: Spectral library matching
The spectra-search subtool computes the similarity between all features in the project-space against a selected spectral database. The spectral database must be imported first using the custom-db tool
sirius --input <dbfiles> custom-db --name <mySpectralDB> --location </some/dir>
sirius --input <input> --project <projectspace> spectra-search --db <mySpectralDB>
formulas: Identifying molecular formulas with SIRIUS (Compound Tool)
One of the primary functions of SIRIUS is identifying the molecular formula of a
measured ion. For this task, SIRIUS provides the formulas tool.
sirius --input <inputFile> --project <projectspace> formulas
Available aliases: trees, formula, sirius
Computing fragmentation trees only
If you already know the correct molecular formula and only need to
compute a fragmentation tree, you can specify the formula using the --formulas option.
SIRIUS will then compute a fragmentation tree exclusively for this molecular formula.
If your input data is in .ms format, the molecular formula might already be included in the file.
The --formulas option also accepts a comma-separated list of candidate molecular formulas.
sirius --input <inputFile> --project <projectspace> formulas --formulas <formula>
Instrument-specific parameters
Datasets vary in mass errors, noise levels, accuracy of isotope pattern intensities depending on instrument type and setup.
By default, SIRIUS uses a profile for Q-TOF data, with a 10 ppm mass deviation. This profile is suitable as a good default profile for a range of instruments.
If you are using data with much lower mass errors, such as Orbitrap or FT-ICR spectra, you may need to adjust the parameters accordingly. Conversely, if your data has higher mass errors, you should adjust the profile and mass deviation settings to match.
You can specify the instrument profile using -p <name>, choosing either qtof (default) or orbitrap. The
orbitrap profile uses a mass deviation of 5 ppm and has slightly different isotope scoring settings.
For FT-ICR data, we recommend using the orbitrap profile and specify an even lower mass deviation.
You can specify the maximum allowed mass deviations for MS1 and MS2 separately:
sirius --input <inputFile> --project <projectspace> formulas -p orbitrap --ppm-max 2 --ppm-max-ms2 5
Large data sets with high mass compounds
When computing molecular formulas with SIRIUS a few high mass compounds usually need most of the computing time, and some of them might not finish computing in reasonable time at all and block your whole analysis.
The most straightforward solution is to exclude high mass compounds from the analysis, by setting a mass threshold. This will usually allow you to annotate the vast majority of your data. However, many of the higher mass compounds will work just fine, and it would be a pity to not annotate them. Therefore, the recommended solution is to set a per-compound timeout (--compound-timeout) so that a few hard cases cannot block your analysis. Read more about How to deal with high mass compounds.
fingerprints: Predicting molecular fingerprints (Compound Tool)
Molecular fingerprints can be predicted using the fingerprints command after calculating molecular formula candidates with the formulas tool.
A fingerprint is generated based for a specific molecular formula candidate and its corresponding fragmentation tree. By default, SIRIUS predicts fingerprints for multiple high-scoring formula candidates by applying a soft threshold on the SIRIUS score.
sirius --input <input> --project <projectspace> formulas fingerprints
Available aliases: fingerprint
classes: Database-free compound classes prediction with CANOPUS (Compound Tool)
The classes tool enables the prediction of compound classes directly from the probabilistic molecular fingerprints generated by CSI:FingerID (using the fingerprints command). One key advantage of CANOPUS is its ability to provide compound class information even for compounds that have no matching hit in a structure database. CANOPUS classes are required for confidence score estimation.
sirius --i <input> --project <projectspace> formulas fingerprints classes
Available aliases: canopus, compound-classes
structures: Identifying molecular structures (Compound Tool)
The structures tool in SIRIUS allows you to search for molecular structures in a structure database
using CSI:FingerID.
To perform structure database search, molecular fingerprints must first be predicted using the fingerprints tool. For improved formula ranking within biologically derived samples (or any other set of derivatives), we recommend to run the zodiac tool beforehand.
You can specify the database for CSI:FingerID to search in, using the --databases option. Available databases include pubchem and bio, among others.
sirius --input <input> --project <projectspace> formulas fingerprints structures --database pubchem
Available aliases: structure-db-search, structure
denovo-structures: Generate de novo molecular structures (Compound Tool)
The denovo-structures subtool in SIRIUS allows you to generate molecular structures de novo from MS/MS data - without relying on any database. To perform de novo structure generation, molecular fingerprints must first be predicted using the fingerprints tool.
sirius --input <input> --project <projectspace> formulas fingerprints denovo-structures
Available aliases: msnovelist
passattuto: Decoy spectra from fragmentation trees (Compound Tool)
The passattuto tool is designed to generate high-quality decoy spectra from
fragmentation trees obtained using the formulas tool. If you’re working with a spectral library,
you can easily create a decoy database.
In SIRIUS 6 passatutto is not available but will be deeply integrated in SIRIUS in the future.
DATASET TOOLS
zodiac: Improve molecular formula identifications (Dataset Tool)
When working with input data derived from biological samples or sets of derivatives,
similarities between different compounds can be used to enhance molecular formula annotations.
ZODIAC leverages these similarities by constructing a network of molecular formula candidates (generated by the formula tool) and re-ranking these candidates using Bayesian statistics (Gibbs Sampling). This approach can reduce the error rate of the top-ranked candidates by approximately 2 fold, with even more significant improvements on challenging datasets containing large compounds.
The zodiac tool is executed after the formulas tool:
sirius --input <input> --project <projectspace> formulas -c 50 zodiac
We recommend to increase the maximum number of
formula candidates (-c) retained after running the formulas tool.
The candidates are the ZODIAC input, and if the correct candidate is missing, ZODIAC cannot
recover it. In order to reduce memory consumption and running time,
ZODIAC dynamically adjusts the number of candidates for each compound based on its m/z. The rationale is that lower-mass compounds are more likely to have the correct molecular formula ranked higher, allowing for fewer candidates to be considered.
By default, ZODIAC uses
10 candidates for compounds with m/z ≤ 300 (--considered-candidates-at-300 10) and
50 candidates for compounds with m/z ≥ 800 (--considered-candidates-at-800 50).
In between, the number of candidates is calculated by interpolation.
The density of the ZODIAC network is primarily determined by two parameters: --edge-threshold
(default: 0.95) and --minLocalConnections (default: 10).
The edge threshold discards 95% of the lowest-scoring edges,
assuming most are incorrect since only one correct candidate exists per compound.
To prevent compounds being disconnected from the rest of the network,
at least one candidate per compound remains connected
to at least
--minLocalConnections other compounds. This introduces an individual edge score threshold for
each compound. Be aware that using --minLocalConnections may requires substantial memory as the entire network is constructed before edge filtering.
For very large datasets, the ZODIAC network might exceed 1TB of system
memory. To manage this, align features across LC-MS/MS runs to reduce the number of compounds. If memory usage is still high,
memory consumption can be dramatically decreased by setting --minLocalConnections=0 to allow filtering low-weight edges during network creation.
However, use this setting with care, since it can result in a poorly connected
network, potentially reducing performance.
sirius --input <input> --project <projectspace> formulas -c 50 zodiac --minLocalConnections 0 --edge-threshold 0.99
Proceed computations and perform recomputations
Compute missing results without recomputing
Assume you have previously computed results for the formulas subtool for compounds with a mass less than 600 Da.
sirius --input <inputFile> --project <projectspace> lcms-align
sirius --project <projectspace> --maxmz 600 formulas
Now, you want to run a workflow that includes formulas, and fingerprints on the same project-space, but without restricting the precursor mass.
sirius --project <projectspace> formulas fingerprints
The formulas results for compounds under 600 Da will be skipped and not recomputed, as these results already exist. Results for compounds with at least 600 Da will be newly generated.
The fingerprints subtool will then be executed for all compounds.
Recompute all results
If you run the same workflow with the --recompute option, all results will be recomputed.
sirius --project <projectspace> --recompute formulas fingerprints
Recompute results for a single subtool
Assume you have a project-space
with complete results for formulas, fingerprints, classes, and structures.
sirius --input <inputFile> --project <projectspace> lcms-align formulas fingerprints classes structures --database pubchem
You now want to recompute the structures results due to incorrect parameters.
sirius --project <projectspace> --recompute structures --database mydb
This
will recompute all structures results without affecting the existing formulas, fingerprints, or classes results.
Note: Recomputing the fingerprints tool results would cause the loss of both structures and classes results.
Proceed with interrupted computations
If a computation is interrupted, simply rerun the same command to resume the process. SIRIUS will skip the computation for existing results and only compute the missing ones.
Special case zodiac:
The zodiac tool operates on the entire dataset rather than individual compounds.
Therefore, if not all compounds have zodiac results, the tool will recompute results for the entire dataset.