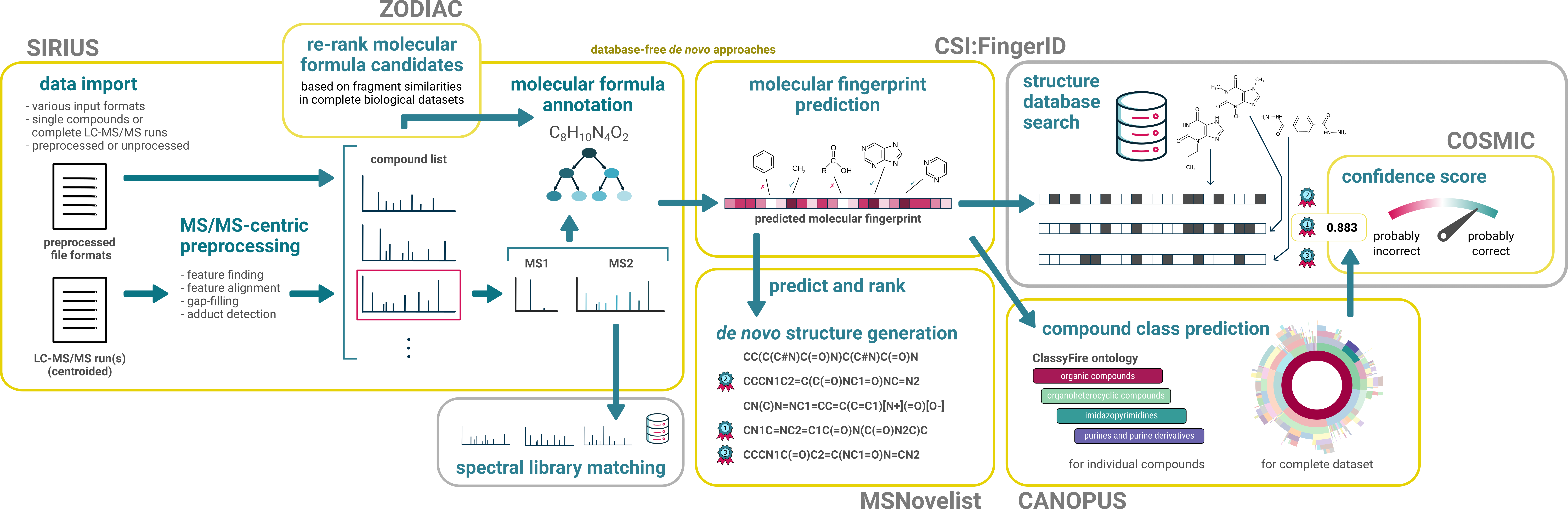
SIRIUS Workflows
SIRIUS is the umbrella application comprising several workflows:
- molecular formula annotation (SIRIUS + ZODIAC),
- fingerprint prediction and compound class prediction (CSI:FingerID + CANOPUS),
- structure database searching (CSI:FingerID + COSMIC),
- de novo structure generation (MSNovelist).
These workflows follow a certain hierarchy, and cannot be freely combined. For example, to predict CANOPUS compound classes, the molecular formula annotation workflow must be run first (or results from a previous run must be available). See the following figure on how the different workflows depend on each other.
Spectral library matching
SIRIUS 6 allows importing local libraries containing spectral reference data. Supported import formats are .ms, .mgf, .msp, .mat, .txt (MassBank), .mb, .json (GNPS, MoNA). Spectra must be annotated with a structure and be centroided.
Spectral library matching is performed using the cosine score with squared peak intensities, ignoring the precursor peak. SIRIUS supports identity search, meaning that library spectra with the same precursor mass (within a given precursor mass deviation) are matched against the query spectrum, and analog search, meaning that library spectra with different precursor masses are matched against the query spectrum. We developed a dynamic programming algorithm that computes the modified cosine similarity for analog search in linear time, to efficiently search datasets with thousands of features against large spectral libraries.
To accelerate searches against large spectral libraries containing hundreds of thousands of spectra, we group spectra by molecular structure and adduct type into clusters. Each cluster is represented by the merged spectrum and a 2D molecular structure. We employ an upper-bound search strategy that estimates the maximum possible (modified) cosine similarity for each cluster. Instead of exhaustively scanning the entire library, we first compute these upper bounds. Only clusters whose estimated upper bound exceeds a predefined threshold then proceed to a full similarity calculation for every single spectrum in the cluster. The cluster representatives (merged spectra) are also treated as valid hits and are listed in all result views (e.g. for substructure annotation).
Spectral matching results improving ZODIAC
ZODIAC uses the top X molecular formula candidates for each molecule to build a similarity network, and uses Bayesian statistics to re-rank those candidates. Spectral library matches with high cosine similarity are set as anchors in the network, meaning the molecular formula is fixed. For analog match we introduce a new node to the network which is also set as anchor.
Spectral matching influence on SIRIUS and CSI:FingerID results
In SIRIUS 6, spectral library matches are added as annotations to CSI:FingerID results and do not influence the ranking of structure candidates. If a high-quality spectral library hit is found for a molecular formula that SIRIUS would not have otherwise considered, that molecular formula is forcibly added to the list of candidates. This ensures that no spectral library matches are overlooked when using CSI:FingerID.
Molecular formula identification with SIRIUS
SIRIUS is the name of the umbrella application, but (for historic reasons) also the name for the identification of the molecular formula. First, a set of candidate molecular formulas is generated. For this set, molecular formula identification is done using isotope pattern analysis on the MS1 data as well as fragmentation tree computation on the MS2 data. The score of a molecular formula candidate is a combination of the isotope pattern score and the fragmentation tree score.
For a deeper understanding of how SIRIUS and ZODIAC work, why they work and how well they work, you can watch the Behind the Scenes talk. Try using SIRIUS in the GUI or CLI.
Generating molecular formula candidates
SIRIUS supports three different approaches for generating the set of candidate molecular formulas for feature annotation:
A thorough understanding of these approaches is crucial for effectively applying the annotation strategy that aligns best with your task or research question.
It is equally important to understand how the molecular formula annotation step affects structure annotation and compound class prediction. Only those molecular formula candidates selected by the annotation strategy are used for structure annotation via database searching and compound class prediction in subsequent steps.
IMPORTANT: Molecular formulas that are not included in the candidate set during this step are excluded from all subsequent processes.
SIRIUS imposes penalties on candidate molecular formulas that significantly deviate from typical biomolecule compositions (e.g., C2H2N12O12 would incur a penalty). However, these penalties are applied cautiously: Only 2.6% of all molecular formulas in PubChem compounds — and thus a small fraction of formulas not categorized as biomolecules — are penalized. SIRIUS never rewards molecular formulas. These penalty rules apply to all approaches.
SIRIUS uses a concise list of outlier molecular formulas that would typically receive penalties under the aforementioned criteria due to their deviation from “biomolecule-like” compositions. These formulas are not penalized, observed in metabolomics experiments (e.g., as solvents), are neither penalized nor rewarded. However, during the computation of fragmentation trees, fragment annotations within MS/MS spectra—subformulas derived from these outlier molecular formulas—may still incur penalties.
De novo annotation
SIRIUS considers all chemically feasible molecular formulas (considering valencies) that match the precursor mass of the molecule/ion: For instance, if your query compound is pinensin A (C96H139N27O30S2, monoisotopic mass of 2213.962 Da), SIRIUS will evaluate all 19,746,670 candidate molecular formulas that match this monoisotopic mass (based on a subset of elements - described below - and 10 ppm mass accuracy).
Considering all molecular formulas implies specifying a set of elements from which these formulas are generated. The elements most abundant in living beings are hydrogen (H), carbon (C), nitrogen (N), oxygen (O), and phosphor (P). This is the default set of elements in SIRIUS. Some less common elements result in very characteristic isotope pattern changes and can be automatically detected from the isotope and fragmentation pattern of the query compound (see Meusel et al.). Detectable elements are sulfur (S), chlorine (Cl), bromine (Br), boron (B) and selenium (Se).
- Default elements: H, C, N, O, P
- Autodetect elements: B, S, Cl, Se, Br
Users should only manually adjust the element set if they have specific prior knowledge about the features of interest. Expanding the element set unnecessarily will result in significantly longer computation times and an increase in incorrect annotations.
This approach can result in molecular formula annotations that may not match any entries in the structure database.
Formula database search
Instead of exploring the entire space of molecular formulas for a given mass and element set, one can alternatively confine the search to a specific database. In that case, SIRIUS exclusively considers molecular formulas included in the chosen database(s), with the option to further narrow down by specific element sets. Naturally, this approach cannot annotate novel molecular formulas (“novel” defined as “not present in the selected database”) and significantly restricts the pool of molecular formulas candidates. As this pool is much smaller than for the de novo method, this approach does not require a predefined element set. The database constrained approach is more likely to annotate formulas with unusual elements that are not recognizable from MS1 than the de novo approach.
Bottom-up search
The “bottom-up” approach serves as a middle ground between the expansive molecular formula space of de novo annotation and the very constrained space of formula database search. This method, inspired by Xing et al., leverages each observed fragment’s mass and corresponding root loss mass from the MS/MS spectrum to query a database of potential subformulas. Pairwise combinations of these resulting subformula candidates for fragments and root losses are then used to construct candidate formulas for the precursor molecule. Thus, the range of precursor formula candidates generated depends on the fragments present in the spectrum. Unlike strict restriction to database entries, this method can detect novel formulas that are combinations of two known molecular formulas. And yet, relying on database entries, this approach generates a much smaller number of candidate formulas compared to de novo annotation, leading to a substantial speed up in computation time.
Given the constrains, strict limitations on an element subset are often unnecessary. The formula database used for bottom-up searches typically includes formulas from the “bio” database along with a list of commonly observed losses. Again, this approach is more likely to annotate formulas with unusual elements that are not recognizable from MS1 than the de novo approach.
Molecular formula annotation strategies
The molecular formula annotations explained above can be utilized either individually or combined. Selecting the appropriate molecular formula annotation strategy is integral for a successful analysis. Below we describe some standard strategies that cover most applications and serve as illustrative examples:
- De novo + bottom-up
We recommend this approach for generic applications.
In the combined approach, features are categorized into “low” (m/z<400) and “high”(m/z>=400) mass features. Bottom-up search is conducted for both categories. For low mass features, SIRIUS also employs de novo molecular formula annotation to ensure comprehensive coverage. This dual strategy minimally impacts (increases) computation times compared to relying solely on bottom-up search. The m/z threshold for categorization can be adjusted to align with runtime constraints and the computational capabilities of your local machine. Element set constraints must be defined for de novo annotation and can optionally be applied to bottom-up search as well. - De novo only
This approach is particularly suitable for discovering “unknown unknowns”.
The “de novo only” strategy should be employed when specifically expecting molecular formulas that cannot be detected by bottom-up search (i.e., the precursor formula is not a combination of database subformulas). The expected element set needs to be well-defined and avoid including too many uncommon elements, as this can lead to a combinatorial explosion of possible candidates for large masses (see the example in de novo). The local machine running the SIRIUS client must be sufficiently powerful to handle de novo annotation of higher mass compounds. - Database search only
This strategy should be employed only when the user is interested exclusively in features that have corresponding entries in a structure database and requires extremely fast computation times.
As the database-only approach will only consider molecular formulas present in the selected databases it will not generate formula annotations without a structure database match. - Bottom-up only
This strategy can be used for a slight speed increase compared to the combined approach.
However, it does not offer significant advantages over the recommended strategy, as the drawbacks of de novo annotation are primarily relevant for high-mass compounds.
Automated lipid detection
Lipid detection (El Gordo) automatically identifies and flags lipid species within complex biological samples.
It runs automatically for every MS/MS spectrum, operating independently of the chosen molecular formula identification method.
Lipid detection analyzes characteristic fragmentation patterns specific to various lipid classes to deduce the lipid’s molecular formula.
Annotations are adhering to standard shorthand nomenclature, such as DG(10:1_8:1) or HexCer(d11:4_16:2).
Identified lipids are flagged in SIRIUS, making them fully searchable and filterable via the user interface filter dialogue.
Once a feature is flagged as a lipid, its molecular formula is automatically fixed, constraining subsequent downstream analysis to biologically relevant candidates.
Be aware that lipid structures are often highly similar, differing only in the position of double bonds. These subtle differences can be indistinguishable using mass spectrometry alone. Thus, annotations reflect the composition but not the exact stereochemistry. SIRIUS also comes with a KMD plot to find related species, including the ones that only have MS1 - but it does not automatically flag those.
The following lipid classes can be detected:
Monoradylglycerol, Diradylglycerol, Triradylglycerol, Betaine diradylglycerol, Betaine monoradylglycerol, Glycosyldiradylglycerol, Glycosyldiradylglycerol, Sulfoquinovosyldiacylglycerol, Sulfoquinovosylmonoacylglycerol, Diacylglycerophosphocholine, Monoacylglycerophosphocholine, Diacylglycerophosphoethanolamine, Monoacylglycerophosphoethanolamine, Diacylglycerophosphoserine, Monoacylglycerophosphoserine, Diacylglycerophosphoglycerol, Monoacylglycerophosphoglycerol, Diacylglycerophosphoinositol, Monoacylglycerophosphoinositol, Diacylglycerophosphate, Monoacylglycerophosphate, Diacylglycerophosphoglycerophosphodiradylglycerol, Ceramide, Hexose Ceramide, Sphingomyeline
Isotope pattern analysis
Isotope patterns of the candidate molecular formulas are simulated starting with the isotopic distributions of the individual elements, and then combining these distributions by folding (see Böcker et al.). The simulated isotope pattern is compared with the measured pattern by assigning probabilities to the observed masses and intensities.
Fragmentation tree computation
Fragmentation trees annotate the fragmentation spectrum with molecular formulas, and identify likely losses between the ions in the fragmentation spectrum. Fragmentation trees can be used to determine the molecular formula of a query compound and to gain insights into its fragmentation. This fragmentation information is utilized in CSI:FingerID to predict the molecular fingerprint of the query compound.
Fragmentation trees are computed directly from the fragmentation spectrum without the need for spectral libraries or molecular structure databases (for the subtle “exemptions” from this rule, see Böcker & Dührkop). Fragmentation trees are computed by combinatorial optimization; the underlying optimization problem constitutes a Maximum Aposterior Estimator. The optimization problem (finding a maximum colorful subtree) is NP-hard but nevertheless solved optimally, explaining why computations sometimes require significant running time for large molecules with rich fragmentation spectra.
With SIRIUS 4.0, fragmentation tree computation has been significantly accelerated—approximately 36 times faster than the previous version—thanks to advanced algorithm engineering. If you believe further speed improvements are necessary, we encourage you to cite our papers on swiftly computing fragmentation trees (Dührkop et al.; White et al.; Rauf et al.; Böcker & Rasche). This recognition provides an incentive for us to continue enhancing our work in this area. We stress that the current version of SIRIUS is millions of times faster than the initial version. In fact, the initial version struggled to process more than 15 peaks in the fragmentation spectrum due to excessive running times and memory requirements.
Modeling fragmentation processes as a tree comes with two restrictions: Namely, “pull-ups” and “parallelograms”.
- A pull-up occurs when a fragment is placed too deep in the tree Due to ourcombinatorial optimization, SIRIUS tends to generate deep trees, favoring many small fragmentation steps over fewer larger ones. For example, SIRIUS will prefer three consecutive C2H2 losses over a single C6H6 loss. While this does not affect the accuracy of molecular formula identification, you should keep this side effect in mind when interpreting fragmentation trees.
- Parallelograms are consecutive fragmentation processes that can occur in more than one order: For instance, the precursor ion might lose H2O followed by CO2, but also CO2 followed by H2O. SIRIUS will always choose one order for such fragmentation reactions, as this is the only valid way to model the fragmentation as a tree.
We have integrated support for experimental setups, such as MSE, MSall and All Ion Fragmentation, where isotope peaks and fragment peaks are measured together in the same spectrum. For these experiments, SIRIUS offers a combined isotope and fragmentation pattern analysis. However, be aware that SIRIUS assumes that only a single ion species is fragmented in each spectrum; it does not support the analysis of chimeric spectra, where multiple ion species are present simultaneously.
For Data-Dependent Acquisition (DDA), fragmentation spectra contain isotope patterns, which are disturbed through the mass filter, leading to non-trivial modifications of masses and intensities. Currently, SIRIUS does not use these altered isotope patterns in its analysis. Instead, it flags these peaks and ignores them during the optimization process.
Improved molecular formula identification with ZODIAC
ZODIAC stands for ZODIAC: Organic compound Determination by Integral Assignment of elemental Compositions.
ZODIAC improves the ranking of the formula candidates provided by SIRIUS. Organisms produce related metabolites derived from multiple but limited biosynthetic pathways. For a full LC-MS/MS run that is derived from a biological sample or any other set of derivatives the relation of the metabolites is reflected in their similarity. Those similarities are in turn reflected in joint fragments and losses between the fragmentation trees and can be leveraged to improve molecular formula identification of the individual molecules.
ZODIAC uses the top X molecular formula candidates for each molecule from SIRIUS to build a similarity network, and uses Bayesian statistics to re-rank those candidates. Prior probabilities are derived from fragmentation tree similarity. Finding an optimal solution to the resulting computational problem is NP-hard, therefore Gibbs sampling is used.
For a deeper understanding of how SIRIUS and ZODIAC work, why they work and how well they work, you can watch the Behind the Scenes talk. Try using ZODIAC in the GUI or CLI.
Molecular fingerprint prediction with CSI:FingerID
CSI:FIngerID identifies the structure of a molecule by predicting its molecular fingerprint and using this fingerprint to search in a molecular structure database. For more details, watch the CSI:FingerID behind the scenes. Try using molecular fingerprint prediction in the GUI or CLI.
Molecular fingerprints can be used to encode the structural features of molecules. These fingerprints are typically represented as binary vectors of fixed length, where each bit describes the presence or absence of a particular, fixed molecular property, such as the presence of a particular substructure.
One example is the PubChem CACTVS fingerprint, which consists of 881 bits.Molecular property 121 represents the presence of at least one “unsaturated non-aromatic heteroatom-containing ring size 3”. The properties are usually defined by SMARTS (SMiles ARbitrary Target Specification) strings. For example, molecular property 357 corresponds to the SMARTS string [#6](~[#6])(:c)(:n). This string describes a central carbon atom connected to another carbon atom via any bond, to a third aromatic carbon atom via an aromatic bond, and to an aromatic nitrogen atom via an aromatic bond. For a complete description of the CACTVS fingerprint, refer to the official specification document. We ignore all molecular properties that can be derived from the molecular formula of the query compound (i.e., bits 0 to 114 from PubChem CACTVS).
Given the molecular structure of a compound, we can straighforward compute its molecular fingerprint deterministically using the Chemistry Development Kit (CDK). Heinonen et al. pioneered the idea of predicting a molecular fingerprint from from a compound’s fragmentation spectrum. Prior to their work, only a limited number of hand-selected properties (presence or absence of certain substructures) could be inferred from fragmentation spectra — primarily in the context of GC-MS with Electron Ionization. For an illustrative example of this earlier approach, see Curry & Rumelhart.
Given the fragmentation spectrum and fragmentation tree of a query compound, CSI:FingerID predicts its molecular fingerprint using Machine Learning (linear Support Vector Machines). For detailed technical information, refer to Shen et al. and Dührkop et al.. CSI:FingerID predicts multiple types of molecular fingerprints: CDK Substructure fingerprints, PubChem CACTVS fingerprints, Klekota-Roth fingerprints, FP3 fingerprints, and MACCS fingerprints. In addition, CSI:FingerID predicts Extended Connectivity Fingerprints ECFP2 and ECFP4 that appear sufficiently often in the training data. Different from other fingerprints, ECFP are not encoded via SMARTS matching. Instead, a hash function encodes the neighborhood of each atom in the molecule. In principle, these fingerprints can encode $2^{32} \approx 4.2 \cdot 10^9$ different substructures (molecular properties). In practice, it is possible but very unlikely that two substructures share the same value, due to a hash collision.
CSI:FingerID predicts only those molecular properties that demonstrate reasonable prediction quality, as assessed through cross-validation (F1 at least (0.25), see below). In total, 5,000 molecular properties are predicted by SIRIUS 6.0.
CSI:FingerID not only predicts whether a molecular property is absent (zero) or present (one), but also provides an estimate of the certainty of this prediction. Mathematically speaking, it estimates the posterior probability that the molecular property is present. A high posterior probability (close to one) indicates high confidence that the property is present, while a low probability (close to zero) suggests confidence that the property is absent. Values between 0.1 and 0.9 indicate uncertainty about the presence or absence of the property. These posterior probabilities are estimated using a method by Platt, hence they are referred to as “Platt probabilities”. However, even a high certainty (e.g., 99%) does not guarantee the presence of a molecular property. CSI:FingerID predicts thousands of molecular properties, and at a 99% confidence level, we still expect about 10 incorrect predictions out of 1000. Additionally, the estimation parameters are derived from the training data, so if the query molecules differ significantly from the training data, the estimates may be less accurate. In addition to Platt probabilities, we also report the performance of each molecular property classifier in cross validation: The F1 score is the harmonic mean of precision (fraction of retrieved instances that are relevant) and recall (fraction of relevant instances that have been retrieved). A classifier with an F1 score close to one is generally more reliable than one with a score close to zero. Again, these scores are based on cross-validation from the training data and should be interpreted with caution.
It is crucial to understand that the molecular fingerprint predicted by CSI:FingerID is not inherently connected to a specific structure in an existing molecular structure database. That
means that even if the correct molecular structure is absent from all known structure databases, the predicted fingerprint remains valid
within the limitations of the predictive power of the method. This allows users to hypothesize about the structure of “unknown unknowns” not present in any existing structure database. Use the Predicted Fingerprints view in the Graphical User Interface (GUI) to examine the predicted molecular fingerprint in detail.
Structure database search with CSI:FingerID
By default, SIRIUS searches for molecular structures in a biomolecule structure database. It can also search in the (extremely large) PubChem database or in custom “suspect databases” provided by the user.
-
When searching the PubChem database, SIRIUS utilizes a local copy with precomputed molecular fingerprints. This avoids the impracticality of computing fingerprints for candidate molecules “on the fly”, which would be too time-consuming. The local copy of PubChem is periodically updated, and users can check the date of the latest update in the database dialog.
-
The biomolecule structure database (bioDB) is an aggregation of several structure databases containing small molecules of biological interest, including metabolites and other compounds of biological relevance, natural products, synthetic products with potential bioactivity, and contaminants observed in experiments. This biomolecule structure database consists of roughly the following datbases: HMDB, KNApSAcK, CHEBI, KEGG, HSDB, MaConDa, Biocyc, GNPS, YMDB, Plantcyc, NORMAN, SuperNatural, COCONUT, BloodExposome, TeroMol, LOTUS, FooDB, MiMeDB, LIPIDMAPS and structures from PubChem annotated with MeSH terms or classified under “bio and metabolites”, “drug”, “safety and toxic” or “food”. he exact composition of this database may vary depending on the version of SIRIUS in use.
Try using CSI:FingerID in the GUI or CLI.
Expansive search (structure database search with fallback)
SIRIUS 6 introduces expansive search, which allows for structure database searches with a confidence score-based fallback. Structure database search is conducted within the user-selected databases (“requested databases”), and then additionally for “PubChem”. If the top hit in PubChem has a confidence score at least twice as high as the confidence score of the top hit from the requested databases, the search will be “expanded” and the results from PubChem will be displayed.
Confidence assessment with COSMIC
The COSMIC confidence score, introduced by Hoffmann et al., provides a measure of confidence for CSI:FingerID structure annotations. While the CSI:FingerID score is designed to rank different structure candidates for a single feature, it is not well suited to rank the top hits across several features based on their likelihood of being correct. The COSMIC confidence score adresses this gap by providing a standardized measure similar to False Discovery Rates and q-values: higher confidence scores indicate a higher probability that the annotation is correct. This is particularly useful for high-throughput experiments. It allows for the analysis of all features in a large dataset using CSI:FingerID, with COSMIC evaluating the top-ranked hit for each feature. The most reliable structure annotations can be selected for further analysis. Importantly, COSMIC does not re-rank structure candidates of a particular feature nor does it discard any identifications; it simply provides an additional layer of confidence assessment.
The confidence score is calculated using Support Vector Machines (SVMs) with enforced feature directionality (different SVMs are applied based on the length of the structure candidate list). The resulting score is a Platt-probability estimate and thus, ranging from 0 to 1.0. However, it’s important to note that this score should not be interpreted as a direct probability of correctness. During evaluation, we found that a score of 0.64 approximately corresponded to a 10% FDR. However, this might vary significantly depending on the specific characteristics of your dataset.
Interpretation of COSMIC confidence values: COSMIC confidence scores should be interpreted with caution. It’s crucial to understand that these scores are not probabilities and, therefore, do not have a direct statistical interpretation. When performing large-scale analyses, it’s advisable to focus on the highest-confidence hits (e.g., the top 1/5/10%), generally independent of their specific confidence value.
Correct annotation with low confidence value:
If the database being searched contains multiple highly similar structures with nearly identical fingerprint representations and CSI:FingerID
scores, correct structure annotations might receive low confidence values.
Without prior knowledge of the correct structure, any of these highly similar compounds could be the true structure, leading to a lower confidence score. This is not a limitation of CSI:FingerID
or COSMIC, but rather of mass spectrometry itself, as the MS/MS spectra of these similar structures will appear almost identical. We recommend using the approximate mode described below, which provides a higher confidence score for hits that are highly similar to the true structure.
For a deeper understanding of how COSMIC works, why it works and how well it works, you can watch the Behind the Scenes talk. COSMIC is part of the structure database search in the GUI and CLI.
Confidence score modes
The confidence score can be evaluated in two modes: exact and approximate.
-
The
exactmode addresses the question “Is this exact molecular structure hit the true structure of my unknown compound?”. It provides a confidence level for the precise match of the structure. Inexactmode, the confidence score tends to be low when the top structure candidate and the second-best candidate are highly similar. This is common for well-studied molecules, where multiple derivatives often exist in the structure database. -
The
approximatemode answers the question “Is this structure hit correct or highly similar to the true structure?”. In this context, highly similar means that the hit is just one simple chemical reaction away from the true structure. Theoretical speaking, the hit and the true structure should have a Maximum Common Edge Subgraph (MCES) distance of 2. For example, a hit where only a side group has been repositioned compared to the true structure would still be considered “correct” inapproximatemode. If you find nearly correct hits useful, it is advisable to use theapproximatemode, which provides a higher confidence score for hits that are highly similar to the true structure.
Compound class prediction with CANOPUS
CANOPUS (Dührkop et al.) predicts the presense/absense of more than 2500 compound classes. These classes range from very broad categories, such as “Lipids and lipid-like molecules,” to highly specific ones, like “Phosphatidylethanolamines,” “Thiazolidines,” or “7-alpha-hydroxysteroids.” Most of the compound classes are derived from the ClassyFire ontology. Unlike ClassyFire, however, CANOPUS predicts these classes based solely on MS/MS data and without requiring database information. This means it can identify a class even if no molecular structure of that class exists in the molecular structure database.
It is important to note that CANOPUS’s classification does not adhere to the concept of attributing a compound to its biosynthetic precursor or pathway. Instead, it categorizes compounds based on functional groups and common substructures. In the ClassyFire ontology, every compound belongs to multiple compound classes, which describe structural patterns. For example, a dipeptide is classified as both an amino acid (due to containing an amino acid substructure) and a carboxylic acid (due to containing an carboxylic acid substructure). Similarly, a glycosylated amino acid might belong to the compound classes of amino acids and hexoses.
Different from how compound classes are often described in chemistry textbooks, ClassyFire compound classes do not describe the biosynthetic origin. For example, a phytosteroid might be classified as bile acids in Classyfire, because both class of compounds share the same backbone, although they are involved in different biochemical pathways. Without additional knowledge about the measured organism, the MS/MS spectrum alone cannot determine the biochemical origin of a compound class, as the same compound may be derived from different biosynthetic precursors.
Additionally, CANOPUS predicts compound classes based on the categories from NPClassifier. This classification system is more general, but may aligns better with the concept of biosynthetic pathway mapping. However, it is still not using taxonomic information, relying solely on MS/MS data for its predictions.
Be aware that there is no universally accepted threshold to classify predictions as “good” or “bad.” The posterior probability estimates provided do not adhere to a fixed standard, and while a binary classifier might suggest a threshold of 0.5, this is not always sufficient in real-world applications. If a user desires more nuanced classifications, such as “Yes,” “No,” and “Maybe,” thresholds like 0.15 and 0.85 may be helpful, although these are just approximations. Moreover, for statistical analysis, such as determining the number of occurrences of a specific compound class in a sample, users can sum up the probabilities to get an expected count. It’s also important to consider that when a compound is significantly different from known compounds, CANOPUS may return lower probabilities for all compound classes, which means users might need to accept predictions with probabilities below 0.5 depending on their specific needs. Thus, the choice of threshold depends on the context and the user’s tolerance for uncertainty.
For a deeper understanding of how CANOPUS works, why it works and how well it works, you can watch the CANOPUS behind the Scenes talk. Try using CANOPUS in the GUI or CLI.
De novo structure generation with MSNovelist
MSNovelist (Stravs et al.) generates molecular structures de novo from MS/MS data - without relying on any database. This makes it particularly useful for analyzing poorly represented analyte classes and novel compounds, where traditional database searches may fall short. However, it is not intended to replace database searches altogether, as structural elucidation of small molecules from MS/MS data remains a challenging task, and identifying a structure without database candidates is even more difficult.
MSNovelist generates structures which can serve as a great starting point for elucidation of specific unknowns. MSNovelist generates multiple candidate structures from the predicted molecular fingerprint. These candidates are represented in SMILES format and are sampled using an autoregressive model, which generates each SMILES token by token. Once the candidate structures are generated, they are ranked using CSI:FingerID. The proposed structures can serve as an excellent starting point for the elucidation of specific unknown compounds. This information can be further enriched by CANOPUS compound class predictions, providing a broader context for the identified structures.