With SIRIUS 6 we have transitioned from a file-based project space to a Nitrite database. This shift was essential to enhance performance and enable new features. The CLI, API and GUI now store computed results as SIRIUS project space. The project space can in turn be used as input for the GUI or the CLI. Consequently, results generated through an automated workflow using the CLI or API can be reviewed and accessed through the GUI.
Input
SIRIUS supports multiple MS data formats:
.ms,.mgf,.mat/.msp, and Agilent’s.cef: These formats contain pre-processed peak lists for each feature..mzml, and.mzxml: For these formats, SIRIUS will perform feature detection and alignment. Note that, all data must be centroided.
SIRIUS does not support input of raw file formats. Common workflows to process raw data before using SIRIUS include:
- Use msconvert to convert raw format to
.mzML. Import to SIRIUS. (Preprocessing is handled by SIRIUS.) Watch this tutorial to get more information. - Use MassHunter to convert raw format to
.cef. Import to SIRIUS. (Preprocessing is handled by MassHunter.) - Use mzmine export to SIRIUS to get a
.mgffile. Import to SIRIUS. (Preprocessing is handled by MZmine.) Watch this tutorial to get more information. - Use MS-DIAL export to SIRIUS to get a
.matfile. → Import to SIRIUS. (Preprocessing handled by MS-DIAL.)
Additionally, many vendor tools now offer .mzML export, which is a convenient and widely supported option for use with SIRIUS.
Peak list-based formats
MGF-Format
SIRIUS supports the MGF (Mascot Generic Format), initially developed
for peptide spectra in the Mascot search engine. Each MGF file can contain multiple spectra, each beginning and ending with the BEGIN IONS and END IONS markers, respectively. Peaks are listed as pairs of m/z and intensity
values, separated by whitespaces, with one peak per line. Additional
meta-information can be included as NAME=VALUE pairs. SIRIUS recognizes the
following meta-information:
FEATURE_ID: Is used in the summary files asmappingFeatureIDto map your results back to your input files.PEPMASS: Indicates the measured mass of the ion (e.g., the parent peak)CHARGE: Specifies the charge of the ion. Since SIRIUS supports only singly charged ions, this value can be either 1+ or 1-.MSLEVEL: Indicates the type of spectrum; it should be 1 for MS spectra and 2 for MS/MS spectra. SIRIUS will automatically treat higher values as MS/MS spectra.
This is an example for an MGF file:
BEGIN IONS
FEATURE_ID=21
PEPMASS=438.32382
CHARGE=1+
MSLEVEL=2
185.041199 4034.674316
203.052597 12382.624023
245.063171 50792.085938
275.073975 124088.046875
305.084106 441539.125
335.094238 4754.061035
347.09494 13674.210938
365.105103 55487.472656
END IONS
See also the GNPS database for other examples of MGF files.
If neither NAME nor FEATURE_ID are given in the file (e.g when importing MGF files from MSDIAL), the value of SCANS will be used as the feature identifier.
SIRIUS MS-Format
While the above-mentioned data formats may lack some necessary information for SIRIUS computations, the SIRIUS MS file format addresses this limitation and is very similar to the MGF format. This custom file format, which uses the .ms extension, is designed to ensure that all required data is available for processing.
Each .ms file contains one measured feature, and can include multiple spectra.
Each line contains either:
- a peak (given as m/z and intensity separated by whitespace),
- meta-information (starting with
>followed by the information type, a whitespace and the value) or - a comment
(starting with the
#).
SIRIUS recognizes the following meta-information fields:
>compound: Specifies the name of the measured feature (or a placeholder). This field is mandatory.>feature_id: Is used in the summary files asmappingFeatureIDto map your results back to your input files.>parentmass: Specifies the mass of the parent peak.>formula: Provides the molecular formula of the feature. This information is helpful if the correct molecular formula is already known and you want to compute a fragmentation tree or recalibrate the spectrum.>ion: Denotes the ionization mode. Refer to the ion modes format for details.>charge: Specifies the charge of the ion (1 or -1). This field is redundant if the ion mode is specified.>ms1: Marks the beginning of MS peak data. All peaks after this line are interpreted as MS peaks.>ms2: Marks the beginning of MS/MS peak data. All peaks after this line are interpreted as MS/MS peaks.>collision: Works the same as>ms2but you can specify the collision energy.
An example of an .ms file:
>compound Gentiobiose
>feature_id 13
>formula C12H22O11
>ionization [M+Na]+
>parentmass 365.10544
>ms1
365.10543 85.63
366.10887 11.69
367.11041 2.67
>collision 20
185.041199 4034.674316
203.052597 12382.624023
245.063171 50792.085938
275.073975 124088.046875
305.084106 441539.125
335.094238 4754.061035
347.09494 13674.210938
365.105103 55487.472656
Note: The .ms file format is SIRIUS’ internal format and may evolve as
the software develops. Nevertheless, we strive to maintain backward compatibility whenever possible.
A more detailed and commented example (which is still a work in progress) of an .ms file can be found here.
LCMS Runs
SIRIUS can import full LCMS runs in .mzML and .mzXML formats through its
pre-processing tool.
The LCMS pre-processing performs feature detection and alignment based on MS/MS spectra, as well as adduct detection and assigns ion types with specific adducts or in-source losses to the features.
Both in the GUI and CLI, this is done automatically when you import your data. In the GUI, parameters of the feature detection are estimated from the data. To specify parameters yourself, use the CLI
(LC-align subtool) or API.
Be aware that formula annotation (and thus subsequent tools as well) will additionally consider the “base ionization”. For example, if the preprocessing assigns [M+H2O+K]+, formula annotation will also consider [M+K]+. However, SIRIUS only displays [M+H2O+K]+ as detected adduct for the input data, while [M+K]+ is only part of the results (formula annotation and so on).
Custom databases
SIRIUS allows you to import custom structure databases and spectral libraries. Any spectral library you import also functions as a structure database. This means that when you add a spectral library, SIRIUS can leverage both the spectral information and the associated structural data for its analyses.
- Structure database from single file can be imported from a CSV or TSV file, where
structures are provided in
SMILESformat [1]. You can also provide a databaseidand anamefor the entries. You can import multiple files containing compounds in SMILES format into a single database.
CN1CCCC1C2=C[N+](=CC=C2)[O-] id-01 Nicotin
CN1C=NC2=C1C(=O)N(C(=O)N2C)C id-03 Caffein
CN1CCC2=CC3=C(C=C2C1C4C5=C(C6=C(C=C5)OCO6)C(=O)O4)OCO3 id-05 Bicculine
- Structure databases can also be imported from directories or
.zipfiles containing SDF files. - Spectral libraries can be imported from directories or
.zipfiles. The supported import formats for spectral data are.ms,.mgf,.msp,.mat,.txt(MassBank),.mb,.json(GNPS, MoNA). Spectra must be annotated with a structure and must be centroided.
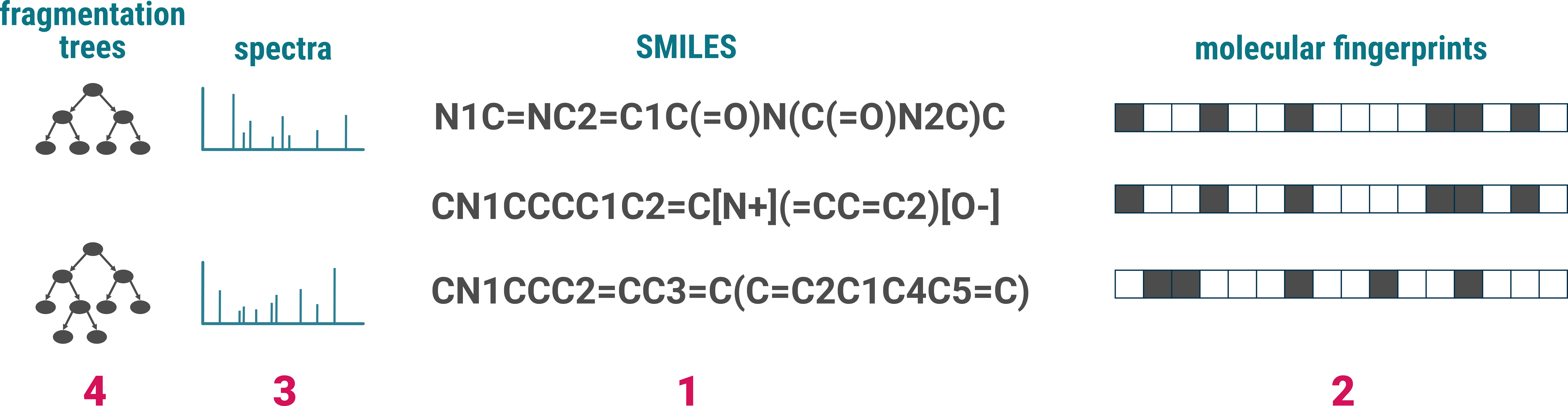
In our machine learning methods, we use PubChem standardized SMILES to represent structures. However, the PubChem standardization is not integrated into the import process. For optimal results, we recommend standardizing your SMILES using the PubChem standardization before importing them. This step is not mandatory, but recommended.
For efficient searching and annotation, SIRIUS generates fingerprints [2] from all imported structures. This enables the software to perform rapid and accurate structure database searches. If a structure is already present in SIRIUS’ internal structure database, the fingerprint will be downloaded automatically. Otherwise, the fingerprint is computed locally on your computer, which may take some time, especially for a large number of structures.
When reference spectra are provided [3], SIRIUS computes fragmentation trees [4] from these spectra. This process allows for efficient substructure annotation, enhancing the utility of the reference data.
SIRIUS offers the option to perform automated computation of bio-transformations during the import process. This feature can be particularly valuable for researchers studying metabolic pathways or drug metabolism, as it helps to identify related compounds within the database.
Output
SIRIUS project space
Since SIRIUS 6, the project space is stored in a singular .sirius file, which is not intended to be human or machine-readable.
This change was implemented to ensure performance for many of the new features and is in no way intended to “close off” results.
You can still generate summaries using the GUI or CLI. To access advanced information or intermediate results (e.g., predicted
fingerprints) you can use the new API.
For robustness, SIRIUS projects require URL-safe names. This means your project name can only contain letters (a-z, A-Z), numbers (0-9), hyphens (-), and underscores (_). Spaces, dots or other special characters are not permitted.
Summary files
The summaries generated by the CLI or GUI are saved in TSV (tab-separated values), CSV (comma-separated values) or XLSX format, or as a zipped TSV (ZIP). You can choose exporting either:
- the top hits only,
- the top hits with adducts (file names end
<file>_adducts.[tsv|csv|xlsx]), - the top
<k>hits (file names end<file>_top-<k>.[tsv|csv|xlsx]), or - all hits (file names end
<file>_all.[tsv|csv|xlsx]).
We recommend exporting the top hits only. Summaries are created separately for molecular formula annotation, spectral library search, structure annotation, compound class prediction, de novo structure identification. When exporting the top hits with adducts, you will get results for the molecular formula annotation only.
You will get the following summary files:
formula_identifications.[tsv|csv|xlsx],spectral_matches.[tsv|csv|xlsx],canopus_formula_summary.[tsv|csv|xlsx],canopus_structure_summary.[tsv|csv|xlsx],structure_identifications.[tsv|csv|xlsx]anddenovo_structure_identifications.[tsv|csv|xlsx].
These files provide convenient access to the results for downstream analysis, data sharing and data visualization. In the summary files, features are sorted by retention time (regardless of the order in GUI). The summaries cannot be imported into SIRIUS but are (re-)created every time a project-space is exported, ensuring that the summaries reflect the most current results.
All summary files contain the mappingFeatureId for each feature. This ID can be used to map your results back to your input data:
- If no feature ID is given in the input file, a fallback
mappingFeatureIdwill be created as<alignedFeatureId>_<INPUT_FILENAME>. - For
.msand.mgffiles, themappingFeatureIDis theFEATURE_IDfrom the input file. - For
.matand.mspfiles, themappingFeatureIDis thePEAKIDfrom the input file. (Note: WhilePEAKIDis not an official field in.mspfiles, it is recognized and will function as expected.) - As
.ceffiles do not support feature IDs, the fallbackmappingFeatureIDwill be used in the summary file. - For data imported from LCMS runs in
.mzMLand.mzXMLformats, the detected aligned features are assigned consecutive numbers, that serve asmappingFeatureID. If you import older project spaces the consecutive numbering might be missing. In that case, the fallbackmappingFeatureIdis displayed as<alignedFeatureId>_UNKNOWN.
All files also contain the overall quality of the feature overallFeatureQuality (GOOD, DECENT, BAD). You can also export a Feature quality summary, containing the basic quality assessment results for all features.
In addition, you can export a ChemVista summary file chemvista_summary.csv
which contains the structure identifications and can be imported directly to the Agilent ChemVista software.
Spectral matches results summary
The summary file spectral_matches.[tsv|csv|xlsx] contains the best spectral hit (highest cosine similarity) for each feature. If you have also conducted analog search, this might also be an analog hit (column analogHit is TRUE).
Molecular formula results summary
The summary file formula_identifications.[tsv|csv|xlsx] contains the top-ranked molecular formula for each feature, based on the SIRIUS score or ZODIAC score, if available.
When multiple adduct candidates correspond to the same precursor ion molecular formula (e.g. [C20H14O6 + NH4]+ and [C20H19NO7 - H2O + H]+),
they will have the identical score.
In such cases, the top-ranked candidate in formula_identifications.[tsv|csv|xlsx] is resolved
to [C20H17NO6 + H]+ only considering the ion type but ignoring adduct types.
The file formula_identifications_adducts.[tsv|csv|xlsx] includes all top-ranked adducts (in this case
[C20H14O6 + NH4]+ and [C20H19NO7 - H2O + H]+). This summary provides additional details, including all scores
displayed in the GUI as well as potential lipid class annotations.
Molecular structure results summary
The summary file structure_identifications.[tsv|csv|xlsx] contains the top-ranked structure result for each feature,
based on the CSI:FingerID score. Note that the molecular formula of this top structure may differ from the
top-ranked molecular formula of this feature. The file includes a column formulaRank, which indicates the
original rank of the molecular formula.
The summary provides the confidence scores for both, exact and approximate modes, as well as CSI:FingerID, ZODIAC and SIRIUS scores.
Additionally, the links column contains reference links to structure databases containing the identified structure.
Compound class prediction results summary
CANOPUS results are reported separately for molecular formula annotations and molecular structure annotations:
- The file
canopus_formula_summary.[tsv|csv|xlsx]contains the predicted compound classes for the top-ranked molecular formula of each feature. - The file
canopus_structure_summary.[tsv|csv|xlsx]contains the predicted compound classes for the molecular formula belonging to the top-ranked structure of each feature.
In both files, the column most specific class indicates the most specific compound class identified for this feature. The columns
level 5, subclass, class, and superclass refer to the ancestors of this most specific class.
In case multiple molecular formulas share the same score (typically due to adducts), we select one molecular formula for each feature, namely the molecular formula for which the CANOPUS probability of the most specific class is highest.
When exporting summaries for the top
De novo structure generation results summary
The summary file denovo_structure_identifications.[tsv|csv|xlsx] contains the top-ranked (based on the CSI:FingerID score) de novo structure generation result for each feature. The file also reports the score from MSNovelist (ModelScore).
Standardized project space summary with mzTab-M
The project space is a SIRIUS-specific format designed to provide comprehensive
access to all results and analysis details. However, it may not be ideal for
sharing data with third-party tools or data archives. To facilitate this, SIRIUS offers an analysis report in
the standardized mzTab-M format (analysis_report.mztab).
The mzTab-M report includes summarized results and links them
to the detailed findings in the corresponding SIRIUS project space.
This ensures that while sharing summarized results using mzTab-M, users retain access to the comprehensive details available in the project space.
Furthermore, SIRIUS transfers meta-information from the input -
such as scan numbers and input data IDs - into the mzTab-M report. This allows
for seamless integration of SIRIUS results with results from other
analyses, such as MS1-based quantification.
Parameter formats
Adducts, losses, ionizations
The nomenclature for losses, adducts, and ionizations is described here.
Molecular formulas
In SIRIUS, molecular formulas must not contain brackets. For instance, use C4H4 instead of 2(C2H2). All molecular
formulas in SIRIUS are treated as neutral. You cannot include charges in a molecular formula. Hence, CH3+ is not a valid molecular formula. Use CH3 instead, and
provide the charge separately.
Chemical alphabets
When specifying a chemical alphabet in SIRIUS, you need to indicate the elements to be considered and their maximum quantities. Chemical alphabets are formatted like molecular formulas, with maximum quantities enclosed in square brackets behind
the element. If no maximum quantity is given, the element might occur
arbitrary often. The standard alphabet is
CHNOP[5]S, which includes the elements Carbon (C), Hydrogen (H), Nitrogen (N), Oxygen (O), and Sulfur (S), with Phosphorus (P) allowed up to five times.